Nov 13, 2015 - 108. (33), were investigated. These strains have previously been ..... Targeted amplicon sequencing (TAS) provides a pipeline for data ...
Look at the following example of a good personal statement whilst referring to the
... person specification for this position together with your reasons for applying.
Bioinformatics exam questions. (a) Describe with one example the difference
between Hamming and Edit distances. [2 marks]. See Lecture 2. The Hamming ...
Apr 20, 2016 - Interaction Using BPSOHS and Emotional Neural Networks. Xiao Wang, Qinke ... use the SNP-SNP interaction in breast cancer susceptibility identification is ... that this novel method is faster than other swarm intelligent algorithms ...
Oct 1, 2013 - *Corresponding author (Email, [email protected]; [email protected]) ..... quence data were used for BLAST search in GenBank data base. ... mL tube containing 10 μL (app 50 ng) of the amplicon, 0.5 μL.
For One-Class Classification problems several methods have been proposed ... In order to train the ν-SVM binary classifier by taking just the positive examples.
Example 1: One Way ANOVA in Excel. A consumer group wants to compare a
new brand of wax (Brand-X) to two leading brands. (Sureglow and Microsheen)
in ...
Jan 1, 2015 - The server responds to all hosts request. Investigate the performance of this network in respect to number of host and packet size. Solution ...
Amplicon contamination in labs masquerades as ... - medRxiv › publication › fulltext › Amplicon... › publication › fulltext › Amplicon...by D Davidi · Cited by 1 · Related articlesDec 15, 2020 — employees who worked in the same research laboratory
EXAMPLE: The solid disc below has radius 4 m and mass 10 kg. Two small objects are placed on top of it. The object on th
Department of Animal Science, Faculty of Agriculture, Bu Ali Sina University, Hamadan, Iran 2. Department of Animal Science, Faculty of Agricultural Science ...
Oct 1, 2013 - [Yanthan M and Misra AK 2013 Amplicon restriction patterns associated with nitrogenase activity of root nodules for selection of superior Myrica.
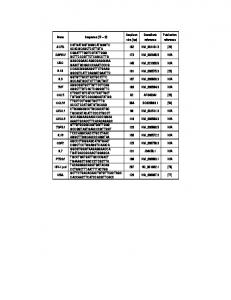
Table 1. Primers list for genomic PCR and RT-PCR. Primer. Primer*. Forward(F)/Reverse(R). Exon(s)*. Amplicon size (bp). Note. Genomic DNA_1.
Oct 23, 2006 - This compressed file contains all scripts required to create a SNP processing pipeline and a web interface for data analysis and visualization ...
Heidelberg, 30. Juli 2013 – Mit der Lösung Kontenplan für ihre Software SNP.
Transformation Backbone® bietet die SNP AG ab sofort eine Anwendung, mit der
sich ... schnell und wirtschaftlich an neue Rahmenbedingungen anpassen.
Xiaoling Wang2, Harold Snieder3,4, Ramasamyiyer Swaminathan5, Keith A. Burling6, ...... Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika.
Oct 12, 2012 - KCNIP4. 4p15. 154. ,3.8610-6. 11.8. **indicates genes in the chromosomal locations where the WTCCC single-SNP analysis showed strong ...
Nov 10, 2010 - Cordell HJ: Epistasis: what it means, what it doesn't mean, and statistical ... Tsai CT, Lai LP, Lin JL, Chiang FT, Hwang JJ, Ritchie MD, Moore ...
May 8, 2014 - Amplicon Library Preparation Methods for High-. Throughput Antibody Sequencing. Ulrike Menzel., Victor Greiff., Tarik A. Khan, Ulrike Haessler ...
Oct 23, 2013 - In addition, Actinomyces europaeus (0.14%) was reported in human abscesses40, Actinomyces neuii (0.03%) was reported to cause ...
reagents used and the available software, cost is less of an obstacle for high-throughput ..... breast cancer. We thank Nicola De Marzo for technical support. This.
Mar 31, 2015 - Allowing politics, training, or other distracting goals to trump service, while representing the mission as âserviceâ. This simply does not describe ...
successfully distinguished class IV SNP (A>T or T>A) in the amplicon where additional ... HRM analysis of DNA amplicons is cost-effective and relatively high ...
Optimized High Resolution (HRM) Analysis Alghoritm Improves Detection Rate of Genetic Variants Jernej Kovač (1), Katarina Trebušak Podkrajšek (1), Tadej Battelino (2)
1. University Medical Centre Ljubljana, Division of Paediatrics, Department for Special Diagnostics 2. University Medical Centre Ljubljana, Division of Paediatrics, Department of Endocrinology, Diabetes and Metabolic Diseases
HRM analysis of DNA amplicons is cost-effective and relatively high throughput screening method that allows detection of genetic variants through dissociation of saturating DNA dyes from denaturing double stranded DNA and consequential loss of fluorescence intensity as function of temperature. The changes in the DNA sequence (SNPs and mutations) cause the change in the melting profile (shape of the meting curve) of the analysed DNA fragment compared to the wild type DNA and the quality of comparison algorithm used to compare those melting profiles is crucial to distinguish between different genetic variants. To improve the detection rate of hard-to-detect genomic variants (SNPs class IV for example), we introduced additional steps to the mathematical analysis and comparison of melting profiles of analysed DNA amplicons. First of all the principal component analysis (PCA) of melting profiles is performed followed by execution of two different unsupervised clustering algorithms – density-based spatial clustering of application with noise (DBSCAN) and expectation maximization (EM) clustering algorithm.
Example of Amplicon with one SNP
Expectation–maximization (EM) algorithm is an iterative method that attempts to find the maximum likelihood estimator of a parameter θ of a parametric probability distribution. The EM iteration alternates between performing an expectation (E) step, which computes the expectation of the log-likelihood evaluated using the current estimate for the parameters, and a maximization (M) step, which computes parameters maximizing the expected log-likelihood found on the E step. These parameter-estimates are then used to determine the distribution of the latent variables in the next E step.
Example of Amplicon with two SNPs The results on the left show the When there are present more comparison of variant calling than one SNP in the analysed with our HRM analysis algorithm amplicon, the melting profiles (a) and HRM v2.0.1 software of the samples become more a (c). Both analysis Experimental workflow complex and the a were performed usanalysis of the reHRM Mastermix (Eva Green Dye) ing data from the sults is rather difDNA sample (10 ng/sample) aligned HRM curves ficult. The pictures Water + PCR primers (b). When there is on the right shows Σvolume= 6 µL only one SNP preresults of the analysent in the amplisis of amplicon with con, the resulting two different SNPs. variant calling is acThere are 9 possible HRM curve capture (ABI 7500 fast RT-PCR System) variants when we b curate with comb mercial software as have two SNPs prewell as our method. sent, but only 4 variPCA + clustering analysis One SNP does not ants are present in pose any obstacle for our analysed popucompared analysis lation. Our analytiDNA sequencing of candidate samples methods and the hocal algorithm (a) as mozygous and hetwell as commercial erozygous variants software (c) both Confirnmation of discovc c of c.47C>T (SOD2 detected 4 different ered genetic variants with genotyping (KBscience, KASp.Val16Ala, rs4880) clusters present in PAR probes) were successfully our population but distinguished. The confidence of the commercial software had variant calling is relatively high troubles with correct clustering >80% (data not shown). of samples (see arrows on pictures a, b & c). Density-based spatial clustering of applications with Principal Component Analysis (PCA) alghoritm is mathematical tool for analysis of multivariate data. It uses orthogonal transformation of set of observations (experimental data) of (possibly) correlated variables into linearly uncorrelated principal components. Each successive calculated principal component describes smaller portion of variance between the datasets. The number of principal components used to describe the variance of our data describes ~95% of variance found in our datasets (data not shown).
Conclusion
noise (DBSCAN) alghoritm is a density-based clustering algorithm because it finds a number of clusters starting from the estimated density distribution of corresponding nodes. The cluster is defined by the parameter of “density reachability”. This means that points that are located in the same cluster are directly density-reachable from each member of assigned cluster. DBSCAN algorithm is defined by two parameters: ε and minimum number of points required to form a cluster (minPts). The parameter ε defines the space in which the algorithm searches for neighbours. If the assigned space is empty the data point is labelled as noise, if the assigned space contains enough neighbours the cluster is formed.
Our HRM analysis algorithm shows improvement with the quality of variant calling compared to commercial software. Moreover our algorithm successfully distinguished class IV SNP (A>T or T>A) in the amplicon where additional SNP was present (data not shown). The demonstrated improvement of variant calling increases the success rate of novel SNP and mutation detection in population studies. We plan to automate the calculation of clustering parameters by introducing additional statistical and software solutions which will further improve variant calling and make analysis more user friendly.