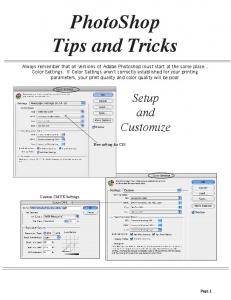
Page 1. Setup and. Customize. PhotoShop. Tips and Tricks. Always remember
that all versions of Adobe Photoshop must start at the same place.
1 Autodesk® AUTOCAD® 2014 Tips and Tricks. Command line. You command
line fans will love the many enhancements, including transparency, autocorrect ...
Access allows you to import data stored in a variety of formats including text and
Microsoft Excel. We recommend you use Microsoft Excel because this format is ...
To enable presenter view when you have two displays (your computer ... Q
PowerPoint 2010 64-bit is not compatible with 32-bit versions of QuickTime or
Flash.
Drill a small hole in the rim and insert a needle through the vinyl tire. Make sure the needle wil fit in the correspond
Features and services may require a Windows Live® ID, network connectivity
and an ... 3 2.1+ EDR. MS office. 3. No. SPECIFICATIONS Windows Quantum.
TIPS AND TRICKS: ... Play Xbox Live and get access to free/exclusive games
right on ...
iPad Tips and Tricks. ➢ The Operating System. The iOS is what powers the iPad's
hands-on features. Review: • Tap to "click" or select something on the screen.
iPad/iPhone Tip. Apple is notorious for their autocorrect technology, which often
ends up correcting words that do not need to be corrected (and sometimes ...
by Sylvain Vervoort aydreaming about trading? Get in a trade early and close to
the low point; stay in as long as needed and get out fast at the top! The problem,.
Garbage or pieces of vehicles are perfect to fill an empty corner. Use some colorful pieces of garbage or equipment to b
Powerful new parametric drawing functionality in AutoCAD 2010 enables you to
dramatically increase productivity by constraining drawing objects based on ...
function sliderDemo(). { var myDiag=new Dialog(); var mySlider=new Slider();.
myDiag.add(mySlider);. mySlider.label="Example Slider". // set min, max and ...
Tips, Tricks, and Tools for Selecting,. Developing, and. Implementing Simple and
Successful Solid. Phase Extraction. Methods. Doug Hanson.
Tips and Tricks in 45 minutes (maybe more :) Olfa Nasraoui, University of Louisville. Tutorial for the Data Science for Social Good. Fellowship 2015 cohort.
Jul 22, 2018 - tions provided by your word processor and type in your ideas in the ...... an appropriate image-processin
Looking to get your Start Button Back? Try Classic Shell. It very easy to use and
free. •. Press the Windows key to enter the tiled Start screen. (or bring up your ...
http://www.howtogeek.com/howto/14529/the-complete-list-of-ipad-tips-tricks-and-
tutorials/. Save Images while Browsing the Web. Want to save an image that ...
Oxford Technology Group. Your IT Solutions Partner! Windows 8 Tips and Tricks.
Windows 8 sold more than 40 million copies and outpaced Windows 7 ...
Advanced Computers Tips and tricks. 4 | Page. 2) Tip : Shutdown Timer trick in
Windows 7. Steps : o Click Start type CMD in the search box. Right click and click
...
Methotrexate Tips & Tricks. For Patients by Patients. MTX is well-studied. MTX has been studied extensively. We know
Apr 2, 2017 - Tricksâ. The best way is to make a short five seconds break after each paragraph and think were you ....
BCEL[2] being the most renown and, probably, most elaborated. It is used by
such tools as Xalan stylesheet compiler[9] and Mercury Interactive's Topaz J2EE
...
iPhone Tips and Tricks #22 - Customize the iPod buttons . ..... The most popular
way to fix a "bricked" iPhone or iPod Touch is by putting it into. DFU mode. This is
tricky, but ... Hold the Home button, then hit the "Power/Sleep" button, the scree
Titel van boek. Plaats, land/Staat: Uitgever. ➢ Refereren hoofdstuk uit
verzamelbundel: Auteur, A. A. & Auteur, B. B. (jaartal). Titel van hoofdstuk. In
C. C..
“A command-line interface (CLI) is a mechanism for interacting with a computer
operating system or software by typing commands to perform specific tasks.
Command-line tools of ChemAxon: tips and tricks
György Pirok
Solutions for Cheminformatics
Command-line interface “A command-line interface (CLI) is a mechanism for interacting with a computer operating system or software by typing commands to perform specific tasks.” (Wikipedia)
msketch The msketch tool starts MarvinSketch applications opening a selected molecule of the file specified as input parameter.
mview caffeine.mol
msketch caffeine.mol
mview The mview tool starts MarvinView application opening the file specified as input parameter.
mview caffeine.mol
mview dsstox.sdf
mview The layout of molecules in mview can be customized by command line parameters. The example below forces to open and SDfile in a matrix view. mview dsstox.sdf --gridbag
It is possible to display only a part of a large file. mview nci.smiles –s 15000 –n 400
The number of displayed columns and rows can be set as parameters as well. mview nci.smiles –c 10 –r 10
These settings are particularly useful when molecules are piped into mview for display as it will be shown later.
molconvert The molconvert script can be used to convert a structure file to an other format. molconvert mrv caffeine.mol –o caffeine.mrv
Merge the molecules of the Molfiles in the current directory to a single SMILES file. The structures are aromatized and explicit hydrogens are removed. molconvert smiles:a-H *.mol –o output.smiles
Merge the SMILES files in the current directory to a single SDfile. The structures are dearomatized, and 2D atom coordinates are calculated. molconvert -2 sdf:-a *.smiles –o output.sdf
molconvert, convert2image Generate jpeg image in 100x150 resolution having yellow background. Many options are available to customize the generated molecule image. molconvert jpeg caffeine.mol –o caffeine.jpg
Batch image generation is possible with the convert2image script that creates a series of numbered images. The script is downloadable from the ChemAxon forum. convert2image "jpeg:w300,h300,mono" molecules.sdf
cxcalc The cxcalc command-line application provides access to plugin functions. The first example below shows general help and lists all available calculations, the second displays calculation specific help. cxcalc -h cxcalc logD -h
Calculated values of a molecule file are usually printed in the form of an indexed table, but the index and table headers can be turned off. cxcalc pKa in.mrv cxcalc -N ih logP in.sdf
cxcalc Enumerate random members of a Markush library. cxcalc -N ih randomMarkushEnumerations -m 50 markush.mrv
Calculate the lowest energy conformer of each molecule of a large file in batch mode and display the results in MarvinView during the calculation. cxcalc lowestEnergyConformer in.smiles | mview --gridbag -
Determine the IUPAC names of the molecules and store them as SDfile fields. cxcalc -S -t NAME -o out.sdf name in.smiles
evaluate The evaluate script provides a command line interface for complex calculations via the Chemical Terms language of ChemAxon. More than a hundred functions can be combined to examine chemical compounds. Determine the number of non-heteroaromatic rings. evaluate –e 'ringCount() – heteroaromaticRingCount()' in.mrv
Calculate an indicator for scaffold hopping. evaluate –e 'dissimilarity("ChemicalFingerprint", actives) - dissimilarity("PharmacophoreFingerprint", actives) > 0.5' in.mrv
Calculate Lipinski's oral drug-likeness flag for each molecule. evaluate –e '(mass() 2' nci.smiles
Find molecules containing carboxyl group having a given pKa value. jcsearch -e "pka('acidic',hm(1)) > 4" -q "[H][O:1]C=[O:2]" nci25k.smiles
standardize Standardizer converts molecules by a list of actions and is usually used as a molecule canonicalization engine integrated with databases. However, the standardize command-line tool provides and easy to use batch conversion interface for files. Merge aromatized molecules of multiple files into a single SMILES file. standardize *.sdf *.mrv -c 'aromatize' –o output.smiles
Remove small components (counterions) and neutralize the remaining molecules. Actions can be listed by two periods as separator. standardize in.mrv -c 'keepone..neutralize' –f mrv –o out.mrv
Convert all nitro groups to the ionic form. standardize in.sdf -c '[O:3]=[N:1]=[O:2]>>[O-:3][N+:1]=[O:2]'
standardize Map reactions by an MCS-based mapping algorithm. standardize in.smiles -c 'mapreaction' –o out.smiles
Convert alias atoms to abbreviated groups by the alias labels and ungroup them after. standardize in.sdf -c 'aliastogroup..sgroups:expand' –f sdf –o out.sdf
Canonicalize molecules acconding to a list of actions specified in an XML configuration file. standardize in.mrv -c config.xml –f sdf –o out.sdf
Retrieve core ring systems using two transforms and pipe the results to MarvinView for display. standardize in.smiles -c '[*R0:1]>>..[*:1]!@[*:2]>>[*:2].[*:1]' | mview -
react Generate reaction products from reactants using virtual reactions with the react program can be used to convert a structure file format to another. Reaction is specified as the –r option. $ react -r '[H:2][C:1]=[O:3]>>[H:2][C:1][O:3][H:4]' "O=Cc1ccccc1" OCc1ccccc1
IUPAC and traditional names are handled by ChemAxon tools as a native format providing outstanding usability for chemists. react –r esterification.smarts '2-hydroxybenzoicacid' 'acetic acid' aspirin
Combinatorial libraries can be produced by multimolecular reactions using the reactants from files. react –m comb –r acyl.rxn alcohols.sdf acidhalides.smiles -o esters.smiles
Summary Command-line interfaces are high performance applications and are available for all ChemAxon products. Only few could be mentioned here. They serve as easy to use tools for those who work with structure files and they shine the most when used in batch mode.
Find out more • Product descriptions & links www.chemaxon.com/products.html